Bioinformatics Analysis: A Key Tool in Computational Biology
As computational power increases and becomes more accessible, the age of bioinformatics will accelerate our ability to understand and tackle global challenges like never before. From discovering new antibiotics to fighting pandemics or making agriculture more sustainable, the promise is great and the applications are already rolling in.
But it’s not all about producing new data, when so much already exists. Analysing data is hugely important. Sharing the results of this requires “showing your working”: the data you used, the methods you employed, the software you used (with versions and parameters). This all takes time and effort, and bioinformaticians can help.
Bioinformatics is a field that combines biological knowledge with computer programming and large sets of big data.
Computational biology is a field that uses computer science, statistic, and mathematics to help solve problems in biology.
Molecular docking services
Molecular docking is a key tool in structural molecular biology and computer-assisted drug design. The goal of ligand-protein docking is to predict the predominant binding mode(s) of a ligand with a protein of known three-dimensional structure.
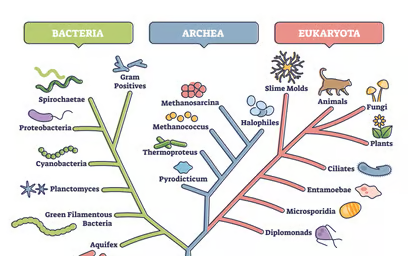
Phylogenetic tree construction and analysis services
Phylogenetic analysis is the study of the evolutionary development of a species or a group of organisms or a particular characteristic of an organism.
A phylogenetic tree (also phylogeny or evolutionary tree) is a branching diagram or a tree showing the evolutionary relationships among various biological species or other entities based upon similarities and differences in their physical or genetic characteristics .
Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the “target” protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein.
Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence.